
Clínica de la enfermedad

Principales hallazgos
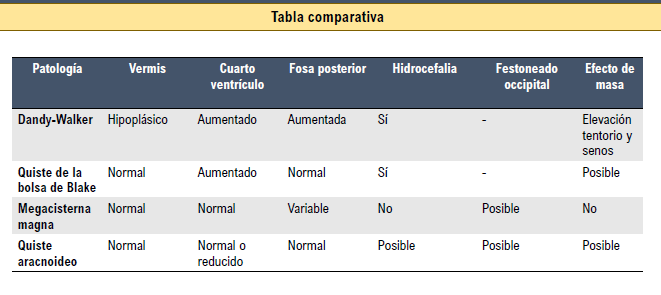
Malformaciones de la fosa posterior:
1. Malformación de Dandy-WaIker:
- La más frecuente dentro de este grupo; suele ser esporádica
- De forma aislada o asociada a otras malformaciones, cromosopatías y trastornos de herencia mendeliana
- Diagnóstico prenatal cada vez más frecuente - Clínica antes del 1º año, con síntomas de HTIC: macrocefalia (90-100%)
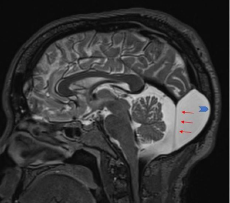
- Alteración en la interacción entre el cerebelo y el mesénquima de la fosa posterior durante el desarrolloSignos
- Hipoplasia del vermis cerebeloso (menos frecuente: agenesia), rotado anterosuperiormente
- Dilatación quística del cuarto ventrículo (no es espacio subaracnoideo)- Otros hallazgos: fosa posterior agrandada; elevación del tentorio, tórcula y senos transversos, hidrocefalia, disgenesas del cuerpo calloso...
2. Hipoplasia aislada de vermis inferior:
- Ausencia parcial de la porción inferior del vermis cerebeloso
- El remanente del vermis así como los hemisferios cerebelosos, el cuarto ventrículo y la fosa posterior son de características normales. Resultados funcionales favorables a largo plazo
- Muchos de los casos descritos en la literatura como “variantes Dandy-Walker” corresponden a esta entidad
3. Quiste de la bolsa de Blake:
- Defecto esporádico secundario a un fallo en la regresión y permeabilización de la bolsa de Blake: no se forma el agujero de Magendie, ausencia de comunicación entre 4º ventrículo - espacio subaracnoideo, con hidrocefalia secundaria
- Hidrocefalia y macrocefalia en período neonatalSignos
- Imagen quística extraaxial de localización infra- y retrocerebelosa, que se corresponde con un “divertículo” del cuarto ventrículo dilatado
- Hidrocefalia es un hallazgo constante (diferencia con megacisterna magna)
- Vermis ligeramente desplazado, con aplanamiento de su porción inferior, pero sin hipoplasia. Fosa posterior de tamaño normal
4. Megacisterna magna:
- Cisterna magna de > 10 mm en cortes sagitales: agrandamiento focal del espacio subaracnoideo en las porciones posterior e inferior de la fosa posterior. Comunica libremente con el cuarto ventrículo y el espacio subaracnoideo
- Retraso en la fenestración de la bolsa de Blake
- Variante anatómica de la normalidad (hallazgo incidental): vermis y cuarto ventrículo normales, sin hidrocefalia ni efecto masa
5. Quiste aracnoideo de la fosa posterior:
- Se forman por separación / duplicación de la membrana aracnoidea
- No comunican con el sistema ventricular ni espacio subaracnoideo
- 10% en fosa posterior, de localización variable
- Pueden aumentar de tamaño por sangrado, secreción de líquido por su pared o cambios osmóticos
- Hallazgo incidental o clínica por efecto masa (hidrocefalia obstructiva, festoneado occipital)
6. Rombencefalosinapsis:
- Ausencia parcial o completa del vermis: fusión de hemisferios cerebelosos, núcleos dentados y pedúnculos cerebelosos superiores
- Esporádico, mayoría no sindrómicos. Puede aparecer en síndrome VACTERL o Gómez-López-Hernández
- Puede asociar hidrocefalia (por estenosis del acueducto) y anomalías supratentoriales (ausencia de bulbos olfatorios, agenesia CC)
Malformaciones del desarrollo cortical cerebral:
• Proliferación celular (2º-4º mes EG): neuronas y células gliales se desarrollan desde sus precursores neuroblásticos en la región ventricular y subventricular
➢ Anomalías por exceso o déficit de neuronas (macro o microcefalia)
➢ Anomalías por proliferación anormal
1. No neoplásica: hemimegalencefalia, displasia cortical con células balonadas, hamartomas corticales de la esclerosis tuberosa
2. Neoplásica: tumor neuroepitelial disembrioplásico (DNET), ganglioglioma, gangliocitoma
• Migración neuronal (3º-5º mes EG): desde la placa germinal hasta la superficie pial estructurándose en seis capas sucesivas (las primeras en migrar ocupan la parte más profunda de la corteza)
➢ Alteraciones por déficit/exceso de migración: lisencefalia, heterotopia en banda, espectro córtex empedrado
➢ Migración ectópica: heterotopias
• Organización cortical (22 semanas gestación – 2 años edad): diferenciación celular, formación del patrón arquitectónico cortical normal
➢ Alteraciones en la formación de las circunvoluciones y en la organización cortical laminar normal: polimicrogiria, esquisencefalia, displasia cortical sin células balonadas
1. Displasia cortical focal:
Grupo heterogéneo de lesiones caracterizadas por “dislaminación” o por desorganización de la citoarquitectura cortical normal- Engrosamiento cortical focal con pérdida de la diferenciación sustancia blanca-gris
- Imagen lineal o curvilínea de intensidad de señal anormal que puede extenderse desde la unión corticosubcortical hasta la sustancia blanca subyacente
- Si se extiende hacia el VL: “transmantle dysplasia” (típico de la tipo 2)
- Macrogiria y mayor profundidad de surcos en la región de DCF
- Diagnóstico diferencial con gliomas
2. Hemimegalencefalia:
- Hiperplasia parcial o total asimétrica de un hemisferio cerebral, ya sea por proliferación aumentada o apoptosis disminuida
- Puede afectar al cerebelo / tronco del encéfalo ipsilateral
- Asocia diferentes grados de polimicrogiria, paquigiria, heterotopia o lisencefalia. Aislada o dentro de otros síndromes malformativos
- Clínica: RN con macrocefalia; convulsiones, hemiplejía, retraso en el desarrollo
3. Lisencefalia:
- Grupo de malformaciones por anomalías en la migración celular, caracterizadas por un número reducido de circunvoluciones. Espectro lisencefalia-paquigiria
- Cinco categorías: lisencefalia clásica, “en empedrado” (cobblestone), ligada a X con agenesia del CC, con hipoplasia cerebelosa, y microlisencefalia
- Clínica: epilepsia refractaria, retraso en el desarrollo, hipotonía, microcefalia
4. Heterotopia:
- Sustancia gris de ubicación anómala, por fallos en la migración de las neuronas desde la matriz germinal hasta la corteza
- Clínica: epilepsia
- Clasificación:
A) Heterotopia periventricular o subependimaria:
• Alteración al inicio de la migración, cerca de la pared ventricular• Puede darse en la esclerosis tuberosa
B) Heterotopia subcortical:
• Grupos de neuronas en sustancia blanca profunda o subcortical (lesiones adyacentes a
ventrículos o al córtex)
• Forma nodular, curvilínea o mass-like
• Hemisferio afectado de menor volumen, con distorsión de la morfología ventricular y
alteración del patrón de surcosC) Heterotopia laminar o en banda:
• Subtipo de subcortical por mutación del gen de la doblecortina
• Capa de sustancia gris separada de la corteza cerebral por una fina capa de sustancia blanca: apariencia de doble corteza
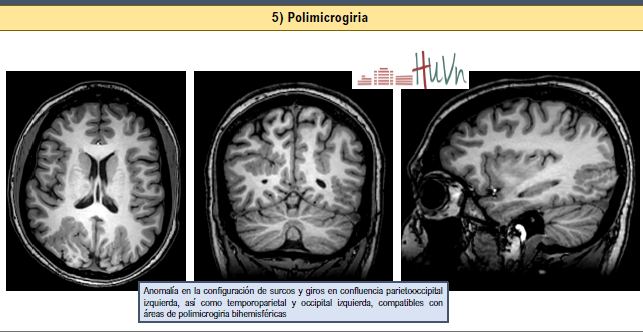
5. Polimicrogiria:
- Circunvoluciones muy finas y onduladas, aumentadas en número, por organización anómala de las seis capas de la corteza cerebral
- Secundaria a noxa intrauterina (infección -CMV-, isquemia, toxina) o anomalías cromosómicas
- Uni- o bilateral, simétrica o asimétrica, focal o difusa. Frecuente asociación con otras de este grupo
- Clínica variada, típicamente convulsiones a los 4-12 años de edad, retraso desarrollo, alteraciones motilidad ocular, parálisis pseudobulbar
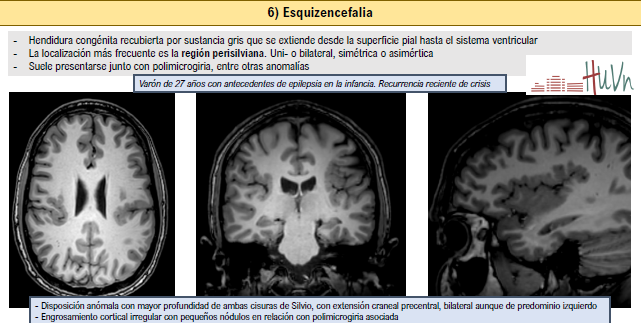
6. Esquizencefalia:
- Hendidura congénita recubierta por sustancia gris que se extiende desde la superficie pial hasta el sistema ventricular
- La localización más frecuente es la región perisilviana. Uni- o bilateral, simétrica o asimértica
- Suele presentarse junto con polimicrogiria, entre otras anomalías
- Esquizencefalia de labio cerrado (tipo I): paredes de la hendidura yuxtapuestas
- Esquizencefalia de labio abierto (tipo II): paredes de la hendidura separadas por LCR
Malformaciones de la inducción ventral
1. Holoprosencefalia: Puede ser una holoprosencefalia alobar sando lugar a un único ventriculo. La semilobar el desarrollo de estructiras cerebrales es mínimo (no separación de los lóbulos frontales anteriores, sí de los occipitales, con diversos grados de desarrollo del resto de estructuras telencefálicas. - No hay septum pellucidum, la cisura interhemisférica está incompleta - Puede haber astas occipitales y temporales de los VL)
2. Displasia septo-óptica
Grupo heterogéneo de trastornos caracterizados por:
- Hipoplasia del nervio / quiasma óptico
- Ausencia de septum pellucidum
- Hipopituitarismo con anomalías hipotálamo-hipofisarias
- Mayoría esporádicos. Por mutaciones genéticas, DG, CMV
- Sospecha clínica: niño con convulsiones, alteraciones visuales y panhipopituitarismo
Otras alteraciones:






