

Clínica de la enfermedad
Síndrome de Apert
Las alteraciones de la bóveda craneal similares a síndrome de Crouzon, junto con alteraciones en manos y pies.
<<acrocefalosindactilia tipo 1>>.
Craneosinóstosis múltiple: La craneosinostosis frontolamboidea es la más frecuente (también bicoronal)-- huesos faciales poco desarrollados--- sindactilias y retraso psicomotor.
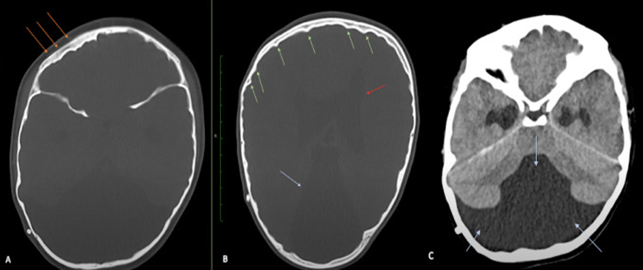
Como ocurre con la mayoría de los bebés con sinostosis bicoronal, la fontanela anterior es grande y está desplazada anteriormente y la sutura metópica permanece abierta, como un mecanismo compensatorio para acomodar el crecimiento del cerebro.
Síndrome de Crouzon
La ausencia de anomalías en manos y pies lo diferencia clínicamente de otros síndromes sinostóticos.
Disostosis craneofacial en la cual las estructuras faciales están subdesarrolladas y la bóveda craneal sufre un cierre primario prematuro de suturas. Destaca la craneosinostosis bicoronal.
Es generalmente familiar (menos severas que en el síndrome de Apert).
Cursa con craneosinóstosis múltiple, hipoplasia del maxilar y afectación de órbitas con exoftalmos, hipertelorismo e hipoplasia de la zona media de la cara.
Hipoplasia del tercio medio facial y la obstrucción de las vías respiratorias superiores que presentan estos individuos.
Microsomía hemifacial
En la actualidad se prefiere óculo-aurículo-vertebral .
Microsomía hemifacial (CFM), acuñado por Gorlin y Pindborg en 1964 (síndrome de primer y segundo arco branquial).
Presentación clínica: Destaca la disostosis otomandibular y asimetría facial. Desde apéndice pre-auricular a deformidad facial extensa con ausencia de pabellón auricular, ausencia de músculos faciales y grave afectación mandibular.
DX: TAC imprescindible (sobre todo CFM tipo II y III)///Modelos 3D mediante estereolitografía en planificación quirúrgica.
/////La ortopantomografía, permite clasificar la CF.